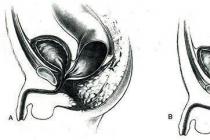
Duchennova myopatia je vrodená patológia, ktorá vyvoláva neustále sa rozvíjajúcu slabosť svalov. Choroba sa začína rozvíjať v detstve. Niekedy rodičia ani nemajú podozrenie, že je dieťa choré: nevšimnú si, že pre dieťa je ťažké bežať, liezť po schodoch a dokonca aj stáť. Pacienti s touto diagnózou by mali byť často vyšetrovaní. Tento typ myopatie postihuje najmä chlapcov, u dievčat je veľmi zriedkavý (sú však prenášačmi).
Duchennova myopatia je vážna patológia, ktorá postihuje svaly ramien, trupu a bokov. Ruky a prsty nie sú ovplyvnené. Problémy vznikajú pri chôdzi, behu. Svalová slabosť sa vyvíja postupne. V detstve, keď sa patológia začína rozvíjať, sú príznaky slabé. S vekom sa prejav symptómov u dieťaťa zintenzívňuje, kvalita života sa zhoršuje. Patológia je diagnostikovaná u jedného chlapca z 3 500 pôrodov.
Patogenéza ochorenia: vo svaloch každého človeka je špeciálny proteín nazývaný dystrofín. U chorých ľudí je veľmi málo bielkovín - kvôli tomu svaly začnú časom ochabovať. Dôvodom vývoja ochorenia je genetická predispozícia: abnormálny gén sa prenáša zo staršej generácie na mladšiu. Príčinou ochorenia môže byť génová mutácia, ku ktorej došlo počas puberty. Potom môžu mať zdraví rodičia dieťa s patológiou.
Existuje 50% šanca, že matka prenášačka prenesie defektný gén na svojho syna. Ak má žena dcéru, dievča sa rovnako pravdepodobne stane nosičom.
Etiológia
Choroba genetickej povahy, kedy v tele pôsobí gén Duchenne.
Existujú určité faktory, ktoré môžu ovplyvniť vývoj patológie:
- genetická predispozícia;
- manželstvo medzi blízkymi príbuznými;
- dedičnosť patológie od matky (najčastejšie);
- mutácia génov počas vývoja plodu;
- anomália v štruktúre chromozómu;
- závažné poškodenie dystrofínu;
- patologické biochemické procesy;
- náhrada spojivového alebo tukového tkaniva svalových vlákien.
Duchennova myopatia sa vyvíja v dôsledku systémových ochorení spojivového tkaniva.
Symptómy
Príznaky Duchennovej choroby sa začínajú objavovať už vo veku troch rokov. Počas tohto obdobia existujú znaky, ktorým by rodičia mali venovať pozornosť:
- pre dieťa je ťažké chodiť, stáť, behať, stúpať po schodoch, chôdza je neistá, dieťa sa kolíše;
- trochu staršie dieťa sa opiera o ruky, aby sa postavilo;
- dieťa zaostáva v učení v porovnaní s rovesníkmi.
V niektorých prípadoch môže byť myopatia odhalená týmto znakom: dieťa zle rozvíja reč. Diagnostika ochorenia zvyčajne nie je náročná, pretože Duchennova myopatia má veľmi špecifické príznaky.
Diagnostika
Pri kontakte s lekárom prvá vec, ktorú lekár urobí, je pozorovanie. Pediatr sa pozrie na to, ako dieťa chodí, behá, vstáva z podlahy. Ak má lekár podozrenie na rozvoj myopatie, nariadi sa špeciálny krvný test.
Testy na diagnostiku Duchennovej choroby:
- Analýza kreatínkinázy (enzýmu). Ak je jeho hladina mnohonásobne vyššia ako normálne, vykoná sa príslušná diagnóza. Ak je indikátor normálny, myopatia Duchenne je vylúčená - lekár hľadá iné príčiny patologického stavu.
- Vykonáva sa biopsia svalového tkaniva. Manipulácia sa vykonáva v celkovej anestézii - dieťaťu sa odoberie malý fragment svalových vlákien na ďalší výskum. Vďaka analýze sa objasní hladina dystrofínu a stav svalového tkaniva.
- Genetické testovanie. Na tento účel sa odoberá krv. V budúcnosti sa identifikujú gény, ktoré ovplyvnili vývoj vrodenej patológie.
Ochorenie sa diagnostikuje hlavne pomocou poslednej uvedenej metódy.
Liečba
V súčasnosti neexistuje žiadny liek na Duchennovú svalovú dystrofiu. Existuje určitá terapia, ktorá pomáha zmierniť stav pacienta. Proces vývoja ochorenia, v závislosti od priebehu liekov, sa v budúcnosti trochu spomalí.
Spôsob liečby Duchennovej myopatie sa vyberá v závislosti od vekovej skupiny. Existujú prípady, keď by sa malo kombinovať niekoľko metód terapie.
Chorý v juniorke predškolskom veku zvyčajne nie je potrebná žiadna špeciálna liečba. Lekár môže rodičom odporučiť:
- rady ako dať fyzické cvičenie choré dieťa;
- genetická konzultácia - ak chcú rodičia vedieť, či sú prenášači (musí to urobiť pre tých, ktorí budú mať viac detí).
V tomto veku musí byť dieťa neustále vyšetrované odborníkom, čo pomôže začať včasnú liečbu.
Spôsoby liečby detí vo veku 5 až 8 rokov:
- dieťaťu sa odporúča použiť podporu svalov nôh – členkovú dlahu, ktorá sa nosí len v noci, alebo dlhé dlahy na predkolenie;
- predpísať užívanie kortikosteroidov - lieky trochu spomalia vývoj ochorenia, svaly budú silné, ale takéto lieky spôsobujú vážne komplikácie, takže dieťa by malo byť pod dohľadom lekára.
Po 8 rokoch sa svaly dieťaťa s Duchennovou svalovou dystrofiou veľmi ochabnú. Je pre neho ťažké chodiť, dieťa sa začína pohybovať na invalidnom vozíku (zvyčajne od 9 do 11 rokov). Pacienti, ktorí začali brať lieky skoro, chodia o niečo dlhšie.
Potom, čo dieťa už nevstáva z invalidného vozíka, vznikajú ďalšie patológie. V tomto čase sú návštevy lekára čoraz častejšie. Čím skôr sa začne s liečbou identifikovanej choroby, tým bude výsledok pozitívnejší.
Rodičia v tomto období by mali pomôcť dieťaťu pohybovať sa na stoličke a vybaviť dom na pohodlný pohyb na invalidnom vozíku.
Ako dieťa rastie, svalová slabosť je čoraz nepríjemnejšia. Človek často potrebuje pomoc príbuzných, dochádza k infekciám pľúc.
Možné komplikácie
Najzávažnejšou komplikáciou je skrátená dĺžka života. V dôsledku rýchleho vývoja ochorenia ochabuje svalstvo, objavujú sa závažné ochorenia srdca. Dýchací systém je narušený.
Ak sa myodystrofia typu Duchenne Becker zistí v počiatočných štádiách vývoja a správne sa zvolí spôsob liečby, pacient môže žiť až 30 rokov.
Súbežne s touto chorobou sa vyvíjajú komplikácie:
- ochorenia chrbtice a kĺbov;
- problémy s gastrointestinálnym traktom;
- porušenie v práci dýchacieho systému;
- srdcovo-cievne ochorenia.
Prevencia
Preventívne opatrenia neexistujú, pretože choroba je dedičná.
Budúci rodičia by sa mali pred plánovaním tehotenstva poradiť s genetikom. To platí najmä pre tie páry, ktoré majú v anamnéze prípady takýchto chorôb.
Rodičia musia byť pozorní k dieťaťu a pri prvých príznakoch, ktoré naznačujú vývoj svalovej slabosti, okamžite vyhľadajte lekársku pomoc na klinike v mieste bydliska.
Aby sa maximalizoval život pacienta s DMD, je nevyhnutné plne dodržiavať všetky odporúčania lekárov, systematicky absolvovať lekárske vyšetrenie a posilniť imunitný systém.
Duchennova (–Griesinger) (progresívna) svalová dystrofia
Synonymum: Merionova choroba.
Dedičné nervovosvalové ochorenie zo skupiny progresívnych svalových dystrofií. Najbežnejšia forma progresívnej svalovej dystrofie viazanej na X. Ďalšou najbežnejšou formou je .
Gén choroby je mapovaný na krátkom ramene chromozómu X (lokus Xp21) a dedí sa recesívnym spôsobom. Podstata X-viazaného recesívneho typu dedičnosti je vysvetlená v článku. Poskytuje tiež informácie o dystrofínovom géne (DYS), štrukturálnych vlastnostiach a funkciách jeho produktu – dystrofínového proteínu a dystrofinopatiách.
Výskyt progresívnej Duchennovej svalovej dystrofie sa pohybuje od 9,7 do 32,6 na 100 000 živonarodených mužov. Vysoká prevalencia ochorenia v populácii je do značnej miery spôsobená vysokou frekvenciou nových mutácií génu dystrofínu, najväčšieho známeho ľudského génu. Priemerná dĺžka života je 25 rokov.
U dievčat je extrémne zriedkavé ochorenie. Môže to mať niekoľko dôvodov: delécia na jednom z chromozómov X zahŕňajúcich lokus Xp21; preskupenie chromozómu X zahŕňajúce Xp21, príp úplná absencia jeden z chromozómov X (napríklad s (Shereshevsky–) Turnerovým syndrómom); uniparentálna dizómia chromozómu X; komplexná heterozygotnosť pre dve mutácie génu DYS; nenáhodná inaktivácia chromozómu X.
V patogenéze ochorenia okrem priameho defektu dystrofínu zohrávajú úlohu aj imunopatologické mechanizmy. Pacienti majú chronické zápalový proces a narušenie regeneračných procesov. Reakcie zápalovej kaskády sa spúšťajú krátko po narodení a sú spôsobené zvýšením obsahu „zápalových“ génových zhlukov vo veku 8-10 mesiacov. V dôsledku defektu lipidovej dvojvrstvy sarkolemy sa zvyšuje jej priepustnosť, najmä pri svalovom napätí; Prispieva aj cytotoxicita makrofágov, ktoré lyzujú sarkolemu po fyzickej námahe (na membráne dystrofických vlákien sa koncentrujú antigény I. triedy hlavného histokompatibilného komplexu (HLA), čo ju robí zraniteľnejšou voči ataku sprostredkovanému T-bunkami). V tomto prípade sa intracelulárny CK uvoľňuje do krvi a extracelulárny vápnik sa ponáhľa do myocytov. Zápalová kaskáda aktivuje produkciu fibrotizujúceho cytokín transformujúceho rastového faktora (TGF-β1), ktorý spôsobuje stratu svalového tkaniva v dôsledku narušených regeneračných procesov. Predpokladá sa, že regeneračná kapacita svalového tkaniva je vyčerpaná v dôsledku nedostatku satelitných buniek po nepretržitých cykloch degenerácie-regenerácie.
Ochorenie je charakterizované pomerne stereotypným priebehom, ktorý prechádza niekoľkými štádiami svojho vývoja, čo je schematicky znázornené na obrázku nižšie.
Typická progresia symptómov s vekom pri Duchennovej myodystrofii (zdroj: http://www.prosensa.eu/img/timeline-DMD-patient.png, upravené)
Pri narodení sa u pacientov s DMD spravidla nezistia žiadne významné abnormality. V prvých mesiacoch je motorický vývoj detí celkom normálny alebo sa vyskytuje s miernym oneskorením. Prvá vec, ktorej ľudia v okolí venujú pozornosť, je oneskorenie začiatku chôdze: takéto deti začínajú chodiť spravidla najskôr 18 mesiacov. Zvyčajne vo veku 4-5 rokov sa porucha chôdze zreteľne prejavuje: nadobúda charakter „kačice“, pacient široko roztiahne nohy, pohybuje sa po špičkách a vzniká bedrová hyperlordóza (tzv. generálska chôdza).

Duchennova svalová dystrofia: charakteristická poloha v stoji, súrodenci vo veku 4 a 6 rokov (zdroj: http://www.jaaos.org/content/10/2/138/F2/graphic-7.medium.gif)
Kolísavá chôdza je spôsobená narastajúcou slabosťou sedacieho a stredného svalu, čo pacientom znemožňuje udržať polohu tela pri prenášaní váhy na jednu nohu. Hyperlordóza je spôsobená slabosťou extenzorov bedrového kĺbu, čo vedie k predklonu panvy a kompenzačnej hyperextenzii v driekovej chrbtici. Pretože je ľahšie udržať telo vzpriamene s equinus nohou, dieťa začína chodiť po špičkách, hoci sa ešte nevyvinuli klinicky významné kontraktúry Achillových šliach.

Schematické znázornenie svalových skupín zapojených do procesu pri Duchennovej myodystrofii (zdroj: http://mda.org/sites/default/files/bmd_dmd_1.jpg)
V priebehu času sa zmeny v chôdzi zhoršujú: ťažkosti sa objavujú pri lezení po schodoch, behu, dieťa začína padať (bez zakopnutia alebo zatmenia) v dôsledku skutočnosti, že „nohy akoby vyšli spod neho“. Typickými sťažnosťami rodičov sú chodenie detí po špičkách a časté pády.
Oneskorenie v rýchlosti motorického vývoja sa často zistí spätne pri analýze anamnestických informácií. Skoré príznaky sa vynárajú nepozorovane. Nedostatočná, v porovnaní s rovesníkmi, pohyblivosť dieťaťa, jeho pohybová pasivita sa často pripisuje vlastnostiam temperamentu a charakteru.Pridávajú sa ťažkosti pri vstávaní zo sedu, pacient je nútený uchýliť sa. Zatiaľ čo pacienti si zachovávajú schopnosť chodiť, ich deformity sú minimálne vyjadrené - je možné len zhrubnutie Achillových šliach a iliopsoas, ako aj mierna skolióza. V dôsledku porážky svalov panvového pletenca je tiež ťažký prechod z horizontálnej do vertikálnej polohy. Motorické funkcie sa zdajú byť relatívne stabilné medzi 3. a 6. rokom života. Zapojenie svalov ramenného pletenca v počiatočných štádiách zvyčajne nie je výrazné, okrem kontroly svalovej sily sa odhalí slabosť proximálnych ramien. Tá však spolu s atrofiou distálnej končatiny a skoliózou začína rýchlo postupovať po preložení pacienta na invalidný vozík. K tomuto zlomu v priebehu ochorenia zvyčajne dochádza medzi 7. a 13. rokom života pacienta, a ak si pacient do 13. roku života zachová schopnosť chodiť, treba predpokladať, že ide o ľahšiu formu X-viazanej progresívnej myodystrofie. -.

Duchennova myodystrofia: charakteristický vzhľad pacienta na invalidnom vozíku (zdroj: http://babylab2.wikispaces.com/file/view/musculardys.jpg/219090918/musculardys.jpg)
Poškodenie svalov ramenného pletenca vedie k obmedzeniu pohybu v ramenných kĺboch. Pacienti nemôžu zdvihnúť ruky nad horizontálnu úroveň, pričom rozsah pohybu v lakťových a zápästných kĺboch a svalová sila zostávajú dlho nedotknuté. Pri pokuse zdvihnúť pacienta pod podpazušie sa zdá, že jeho hlava padá do pliec - príznak "uvoľneného ramenného pletenca". Lopatky zaostávajú za telom - príznakom "pterygoidných lopatiek".

Duchennova svalová dystrofia: symptóm uvoľneného ramenného pletenca (zdroj: http://www.ordodeus.ru/Miopatiya_5.jpg)

Duchennova myodystrofia: symptóm pterygoidných lopatiek (zdroj: http://www.ordodeus.ru/Miopatiya_4.jpg)

Duchennova myodystrofia: atrofia svalov lopatky a ramenného pletenca (zdroj: Suresh Chandran C.J. "Hidden mounts" in Duchenne muscular dystrophy // Neurol. India, 2008. - Vol.56. - N.3. - S.394)
Okrem narastajúcej symetrickej svalovej slabosti v presne definovaných svalových skupinách, po ktorej nasleduje ich, tiež symetrická, atrofia (mimochodom, svalová atrofia je často maskovaná dobre vyvinutým podkožným tukovým tkanivom), sú pre Duchennovú svalovú dystrofiu charakteristické nasledujúce symptómy: :
- Mentálna retardácia . Určité oneskorenie vo vývoji mentálnych funkcií je zaznamenané už v prvých rokoch života. Deti sú bez emócií. Reč sa vyvíja neskoro a je primitívna. Neexistuje žiadne abstraktné myslenie. Zručnosti úhľadnosti a sebaobsluhy sa formujú s ťažkosťami. Mentálna retardácia rôzneho stupňa: od hraničnej intelektuálnej insuficiencie po ťažkú oligofréniu sa u 30 % pacientov s Duchennovou svalovou dystrofiou vyvinie v dôsledku deficitu mozgových izoforiem dystrofínu – apodystrofínu. Závažnosť oligofrénie a porúch vyšších kognitívnych funkcií nekoreluje so závažnosťou svalového defektu a štádiom myodystrofického procesu. Medzi exogénne faktory, ktoré zhoršujú prejavy mentálnej retardácie, patrí rozvíjajúca sa sociálna neprispôsobivosť z dôvodu nemožnosti plnohodnotnej účasti detí v detských kolektívoch (škôlka, škola) pre poruchu motoriky, vplyv nepriaznivých perinatálnych príčin a prípadne mozgová dysgenéza (CT a MRI príležitostne zisťujú príznaky cerebrálnej atrofie). Deti s Duchennovou svalovou dystrofiou majú častejšie poruchy autistického spektra ako je priemer v populácii.
- Zníženie a neskôr strata šľachových reflexov. Najprv sa znížia reflexy kolena, potom zvyšok reflexov. Výnimkou sú Achillove reflexy, ktoré môžu pretrvávať až do neskorších štádií ochorenia.
- Pseudohypertrofia niektorých svalových skupín na pozadí atrofického procesu . Pseudohypertrofia lýtkových svalov je najcharakteristickejšia, hoci sa môže vyvinúť aj v iných svalových skupinách (gluteálne, deltové, brušné svaly a svaly jazyka). Pseudohypertrofia lýtkových svalov vytvára klamlivý dojem o zachovaní svalovej sily a dokonca poteší rodičov. Svalová atrofia môže byť lokálna a generalizovaná. Lokálne sa zisťuje iba v počiatočných štádiách ochorenia, s progresiou patologického procesu sa atrofia stáva generalizovanou až po svalovú kachexiu. Atrofované svaly sú preriedené, pri palpácii ochabnuté.

Duchennova myodystrofia: charakteristický vzhľad pacienta; upozorňuje na pseudohypertrofiu lýtkových svalov (zdroj: http://www.jaaos.org/content/10/2/138/F2/graphic-6.medium.gif)

Duchennova myodystrofia: makroglosia v dôsledku pseudohypertrofie svalov jazyka (zdroj: http://neuromuscular.wustl.edu/pics/people/patients/tonguehypertdmdsm.jpg)
- Zmeny vzhľadu. Pri slabosti a atrofii svalov tváre je nedostatok vrások na čele (príznak "lešteného čela"). Pozoruje sa hypomimia: pacienti nemôžu pevne zavrieť oči, nafúknuť líca, natiahnuť pery do trubice atď. V niektorých prípadoch v dôsledku nahradenia labiálnych svalov spojivovým a tukovým tkanivom pery zhrubnú (pripomína to „ tapírové pery“).

Duchennova myodystrofia: charakteristický vzhľad pacienta z fotografie, ktorú urobil samotný Duchenne (zdroj: http://www.artandmedicine.com/biblio/images/duchenne/DuchenneAlbum16.jpg)
- endokrinné poruchy. V prvom rade k nim patrí obezita. Neuroendokrinné poruchy sa vyskytujú takmer u polovice pacientov. Častejšie ako ostatné sú pozorované,. Vyskytuje sa nízky vzrast.
- Zmeny v kostrovom systéme. Lumbálna hyperlordóza, kyfoskolióza hrudnej chrbtice, deformity hrudníka a chodidiel, difúzna osteoporóza (v dôsledku sedavého spôsobu života, užívania glukokortikosteroidov). Na röntgenových snímkach sa zistí zúženie medulárneho kanála, stenčenie kortikálnej vrstvy diafýzy dlhých tubulárnych kostí.
- Kardiovaskulárne poruchy . Klinicky sa prejavuje labilitou pulzu, krvný tlak , niekedy hluchota tónov a rozšírenie hraníc srdca. Na EKG sa zaznamenávajú zmeny v myokarde (blokáda nôh Hisovho zväzku atď.). Kardiovaskulárny systém sa podieľa na patologickom procese pomerne často a skoro. Asi 73% pacientov má rôzne prejavy srdcovej patológie. Príčinou kardiovaskulárnej patológie je geneticky podmienený nedostatok dystrofínu v kardiomyocytoch. Neexistencia jasnej korelácie medzi závažnosťou poškodenia kostrového svalstva a prítomnosťou závažnej kardiomyopatie u pacientov s Duchennovou svalovou dystrofiou predurčila potrebu venovať osobitnú pozornosť štúdiu markerov zapojenia srdcového svalu do patologického procesu. Ukázalo sa, že delécie génu pre dystrofín nie sú jedinou príčinou poškodenia svalového tkaniva u pacientov s progresívnou Duchennovou svalovou dystrofiou. V súčasnosti vedci identifikujú tri hlavné príčiny: nedostatok dystrofínu v dôsledku genetického defektu; nedostatok glykoproteínu asociovaného s dystrofínom (molekulová hmotnosť 50 kDa) alebo iných proteínov asociovaných s dystrofínom, prítomnosť špeciálneho genetického variantu v štruktúre enzýmu konvertujúceho angiotenzín. Srdcový sval môže byť postihnutý ako v dôsledku všetkých troch príčin, tak aj ich kombinácií. Napríklad nedostatok glykoproteínu spojeného s dystrofínom možno pozorovať výlučne v kardiomyocytoch, zatiaľ čo v tkanive kostrového svalstva bude jeho obsah normálny. Detekcia deficitu proteínov spojených s dystrofínom pri štúdiu biopsie srdcového svalu je prediktorom rozvoja ťažkej kardiomyopatie. V posledných rokoch sa osobitná pozornosť venuje štruktúre enzýmu konvertujúceho angiotenzín: predpokladá sa, že s tým súvisí závažnosť kardiomyopatie. Identifikácia markerov zapojenia srdcového svalu do patologického procesu nám umožňuje odpovedať na mimoriadne dôležitú praktickú otázku - prečo je možné pozorovať kardiomyopatiu u pacientov s miernymi léziami kostrového svalstva, ako aj na možnosť debutu ochorenia s kardiomyopatiou. Počiatočné prejavy srdcovej patológie sa u pacientov spravidla vyskytujú už v ranom veku a postupujú v priebehu rokov. V niektorých prípadoch u detí vo veku 3-5 rokov môžu v klinickom obraze ochorenia dominovať kardiálne symptómy a symptómy svalovej dystrofie môžu byť maskované. Nízka fyzická aktivita pacientov s progresívnou Duchennovou svalovou dystrofiou, relatívne rýchla strata schopnosti samostatnej chôdze, čím sa znižuje záťaž myokardu, ako aj nedostatočná orientácia rodičov na zisťovanie srdcových ťažkostí (hlavná pozornosť je venovaná primárne na poruchy motoriky), vedú k tomu, že menej ako 15 % detí do 14 rokov s poškodením srdcového svalu sa aktívne obráti na kardiológa. Zatiaľ čo podľa cielených štúdií u detí, ktoré nevykazujú srdcové ťažkosti, je poškodenie srdcového svalu zistené u 25 % vo veku do 6 rokov a u 59 % vo veku od 6 do 10 rokov. V budúcnosti sa toto percento znižuje, pretože poškodenie srdca postupuje a deti začínajú mať problémy so srdcom. Patogenéza poškodenia srdcového svalu pri Duchennovej myodystrofii je v súčasnosti prezentovaná nasledovne: progresívna atrofia kardiomyocytov a ich nahradenie fibróznym tkanivom vedie k stenčovaniu myokardu (najmä ľavej komory, ktorá predstavuje hlavnú hemodynamickú záťaž), ako aj k zníženie jeho schopnosti systolickej kontrakcie a diastolickej relaxácie. Ťažká fibróza v oblasti zadných papilárnych svalov vedie k prolapsu cípov mitrálnej chlopne do dutiny ľavej predsiene (prolaps mitrálnej chlopne) s alebo bez mitrálnej regurgitácie. Výskyt prolapsu mitrálnej chlopne u pacientov s progresívnou Duchennovou svalovou dystrofiou sa pohybuje od 25 do 55 %. Zväčšenie veľkosti ľavej predsiene je zvyčajne sekundárne v dôsledku mitrálnej regurgitácie alebo zníženia kontraktility ľavej komory. Srdcové arytmie a poruchy vedenia vzruchu sa vyskytujú v dôsledku progresívnej fibrózy vodivého systému srdca. Poškodenie srdcového svalu sa zvyčajne prvýkrát diagnostikuje medzi 6. a 7. rokom života. S vekom sa frekvencia detekcie srdcových symptómov zvyšuje a vo veku 20 rokov sa patológia kardiovaskulárneho systému vyskytuje u 95% pacientov. Väčšina časté porušenia pozorované u 54 % pacientov boli: tachykardia, arytmie a srdcové zlyhanie. Tieto príznaky sú obzvlášť výrazné v konečných štádiách ochorenia. Vzhľadom na zvláštnosti motorickej aktivity pacientov s Duchennovou myodystrofiou (ako aj s), absenciu srdcových ťažkostí, veľmi malú fyzickú aktivitu, často imobilitu v neskorších štádiách ochorenia, bola v roku 1993 navrhnutá a zavedená do lekárska prax Novým diagnostickým termínom je latentné srdcové zlyhanie.
- Patológia dýchacieho systému . Slabosť dýchacích svalov a bránice spôsobuje zníženie vitálnej kapacity pľúc až o 20 % normy, čo vedie k epizódam nočnej hypoventilácie. Deti často vstávajú so strachom spojeným s pocitom dusenia a boja sa spať. Významný podiel na úmrtnosti má respiračné zlyhanie, ktoré je vyvolané interkurentnými infekciami alebo aspiráciou.
Ochorenie môže prebiehať podľa jedného z niekoľkých klinických variantov, Stručný opis ktoré sú uvedené v tabuľke.
|
|
|
|
|
|
|
|
Stráca schopnosť chodiť vo veku 10-12 rokov. Generalizovaná svalová slabosť, potom skolióza, kontraktúry členku, kolena a iných kĺbov | Telesná hmotnosť je znížená. Duševný vývoj je normálny; kardiomyopatia sa zistí po 8-10 rokoch | ||
|
|
Stráca schopnosť chodiť vo veku 10 rokov alebo skôr. Generalizovaná svalová slabosť, potom skolióza, kontraktúry členku a iných kĺbov | Obezita (mesačná tvár, ukladanie tuku ženského typu). Kardiomyopatia sa zistí po 10 rokoch | ||
|
|
1-2 roky života |
Stráca schopnosť chodiť do 10 rokov, niekedy na 6,5-7 rokov. Skoré mnohopočetné kontraktúry. rýchla progresia | Telesná hmotnosť je znížená alebo normálna. Zhoršená duševná funkcia. Kardiomyopatia sa zistí vo veku 7-10 rokov | |
|
|
Vo veku 6,5-7 rokov sa zisťuje kardiomyopatia s miernymi prejavmi svalovej slabosti (ťažkosti pri výstupe do schodov). Relatívne pomalá progresia | Telesná hmotnosť je znížená alebo normálna. Mentálny vývoj je normálny | ||
|
|
Stráca schopnosť chodiť vo veku 10-12 rokov alebo skôr. Generalizovaná svalová slabosť | Rôzne kombinácie |
Diagnóza Duchennovej svalovej dystrofie je založená na anamnéze (vrátane rodinnej anamnézy), klinickom obraze ochorenia a množstve ďalších výskumných metód. Medzi posledné patria:
Štúdium krvného séra CPK . V zdravých bunkách CPK katalyzuje syntézu kreatínu a ATP z fosfokreatínu a ADP. Pri DMD (a v menšej miere) svalovej dystrofii vedie fyzická aktivita k masívnemu uvoľňovaniu CPK, čo iniciuje zápalovú odpoveď. Preto je hladina CPK v krvnom sére významne zvýšená pri Duchennovej myodystrofii av menšej miere -. Odchýlky v hladine CPK v sére pri niektorých dystrofinopatiách sú uvedené v tabuľke.
|
|
fenotyp |
% prípadov |
koncentrácie CPK |
| Muži | Duchennova myodystrofia |
10-krát alebo viackrát vyššia ako normálne |
|
|
5-krát alebo viac nad normál |
|||
| Dilatačná kardiomyopatia spojená s DMD |
Väčšinou |
Zvýšená |
|
| nositeľky | Duchennova myodystrofia |
2-10 krát vyššia ako normálne |
|
|
2-10 krát vyššia ako normálne |
S vekom koncentrácia CPK postupne klesá, ako resorpcia dystrofických svalových vlákien.
ENMG . Umožňuje odlíšiť primárnu svalovú léziu od neurogénnej. V prvom prípade sa odhalia rýchlo naverbované, krátkodobé polyfázové potenciály motorických jednotiek s nízkou amplitúdou. Ako choroba postupuje, interferenčný obrazec ENMG sa znižuje v dôsledku zníženia náboru a nakoniec sa zaznamená bioelektrické „ticho“ svalu. Údaje ENMG nie sú špecifické a sú rovnaké pre akúkoľvek formu primárnej svalovej lézie. Preto sa v praxi ENMG pri diagnostike Duchennovej myodystrofie používa len zriedka.
Biopsia kostrového svalstva . Vyrába sa na histologické vyšetrenie biopsie, ako aj na imunohistochemické testy a Western blotting. Histologicky v počiatočných štádiách ochorenia sa zisťujú nešpecifické dystrofické zmeny - variabilita veľkosti vlákna, ložiská nekrózy a regenerácie, hyalinizácia; v neskorších štádiách - tukové usadeniny a proliferácia spojivového tkaniva.

Duchennova myodystrofia, časť svalovej biopsie zafarbená hematoxylínom-eozínom. Existuje výrazná fibróza endo- a perimýzia, výrazná variabilita v priemere svalových vlákien v dôsledku prítomnosti atrofovaných a hypertrofovaných vlákien (zdroj: http://img.medscape.com/pi/emed/ckb/pathology/1603817-1607648-1869808-1870086.jpg)

Duchennova myodystrofia, časť svalovej biopsie zafarbená hematoxylínom-eozínom. Dve svalové vlákna, ktoré zaberajú väčšinu úseku, sú v skutočnosti jediné hypertrofované vlákno rozdelené novovytvoreným mostíkom spojivového tkaniva. (zdroj: http://img.medscape.com/pi/emed/ckb/pathology/1603817-1607648-1869808-1870088.jpg)

Duchennova myodystrofia, časť svalovej biopsie zafarbená hematoxylínom-eozínom. V štádiu myofagocytózy je možné vidieť nekrotické svalové vlákno s bledou cytoplazmou. Okrem toho sú na reze viditeľné nasledovné znaky myodystrofického procesu: zvýšenie počtu jadier svalových vlákien, abnormálne vysoká variabilita priemeru svalových vlákien, dve hyalinizované svalové vlákna (bezprostredne pod nekrotickým) a ťažká fibróza

Duchennova myodystrofia, časť svalovej biopsie zafarbená hematoxylínom-eozínom. Niektoré vlákna sa zdajú väčšie, červenkastejšie a „sklenité“. Hovorí sa im „hyalinizované“ alebo „tmavé“ a ide o nadmerne stiahnuté svalové vlákna. Rez tiež ukazuje abnormálnu variabilitu priemeru vlákna, lokalizovanú nekrózu svalových vlákien, mierne zvýšenie počtu jadier svalových vlákien a výraznú endo- a perimyziálnu fibrózu. Treba poznamenať, že hyalinizované vlákna sa môžu vyskytovať ako artefakty. (zdroj: http://img.medscape.com/pi/emed/ckb/pathology/1603817-1607648-1869808-1870089.jpg)

Rez bioptickej vzorky musculus gastrocnemius pacienta, ktorý zomrel na Duchennovú myodystrofiu. Vidno masívne nahradenie svalového tkaniva tukovými bunkami (zdroj: http://upload.wikimedia.org/wikipedia/commons/thumb/4/49/Duchenne-muscular-dystrophy.jpg/800px-Duchenne-muscular-dystrophy.jpg)
Aby sa zvýšila špecifickosť diagnózy, imunohistochemická štúdia so stanovením percenta dystrofínu vo svalových vláknach získaných zo svalovej biopsie farbením antidystrofínovými protilátkami na C-konci, strednej časti a N-konci molekuly dystrofínu. Pri farbení neporušeného svalového tkaniva touto metódou sa odhalí rovnomerná distribúcia farbiva pozdĺž periférie svalových vlákien, čo zodpovedá subsarkolemálnej lokalizácii dystrofínu.

Imunohistochemická analýza biopsie zdravého svalu na dystrofín. Rez zafarbený protilátkami proti dystrofínu ukazuje rovnomerné subsarkolemálne hnedé sfarbenie membrány všetkých svalových vlákien. (zdroj: http://img.medscape.com/pi/emed/ckb/pathology/1603817-1607648-1869808-1870093.jpg)
Pri Duchennovej myodystrofii sa subsarkolemálne zafarbenie nezistí pri použití protilátok na C-, N-koniec a strednú časť (s - iba pri použití protilátok proti N-koncu).

Duchennova myodystrofia, imunohistochemická analýza svalovej biopsie na dystrofín. Žiadne zo svalových vlákien (s výnimkou jediného označeného na obrázku šípkou) nie je zafarbené protilátkami proti dystrofínu. Identický výsledok sa získa pri farbení protilátkami na C-koniec, N-koniec a strednú časť dystrofínu (zdroj: http://img.medscape.com/pi/emed/ckb/pathology/1603817-1607648-1869808-1870095.jpg)
V zriedkavých prípadoch si jednotlivé vlákna môžu počas imunohistochémie zachovať zafarbenie (na predchádzajúcom obrázku je práve takéto vlákno označené šípkou). Môže za to druhá mutácia v géne pre dystrofín, ktorá obnovuje čítací rámec a umožňuje tak syntézu dystrofínu v tomto vlákne. Tieto pozorovania otvárajú vyhliadky na rozvoj jednej z potenciálnych ciest liečby myodystrofie.
Imunohistochemická štúdia vzorky svalovej biopsie je možná aj pre utrofín, autozomálny homológ dystrofínu, ktorý sa syntetizuje v postnatálnom období hlavne v neuromuskulárnych spojeniach. Pri dystrofinopatiách je expresia utrofínu zvýšená a dá sa určiť v sarkoléme.

Imunohistochemická analýza svalovej biopsie na utrofín pri normálnej (A) a pri Duchennovej myodystrofii (B). Obrázok A ukazuje obraz normálnej expresie utrofínu u detí a dospelých: vlákna nie sú zafarbené, utrofín je stanovený len v stene cievy. Obrázok B ukazuje aktiváciu expresie utrofínu pri dystrofinopatii: sarkolemy sú intenzívne zafarbené, čo môže byť odrazom kompenzačnej syntézy utrofínu pri stavoch nedostatku dystrofínu. Oblasti čistenia v cytoplazme svalových vlákien zodpovedajú artefaktom vo forme ľadových mikrokryštálov (zdroj: http://img.medscape.com/pi/emed/ckb/pathology/1603817-1607648-1869808-1870172.jpg)
Western blotting - moderná, vysoko citlivá analytická metóda používaná na detekciu špecifických proteínov v komplexných zmesiach pomocou protilátok. Metóda je založená na kombinácii gélovej elektroforézy a imunochemickej reakcie antigén-protilátka. Pomocou gélovej elektroforézy sa proteíny separujú v polyakrylamidovom géli. Ďalej sa proteíny prenesú na nitrocelulózovú alebo PVDF membránu. Potom sa detegujú pomocou protilátok „sendvičovou“ metódou: najprv sa proteíny naviažu na primárne (mono- alebo polyklonálne) protilátky, ktoré sa následne naviažu na sekundárne protilátky konjugované s enzýmami (chrenová peroxidáza alebo alkalická fosfatáza). Vizualizácia študovaného proteínu sa dosiahne vykonaním vhodnej biochemickej reakcie za vzniku produktu, ktorý sa stanoví kolorimetrickými, chemiluminiscenčnými, fluorescenčnými detekčnými metódami. Množstvo proteínu možno odhadnúť pomocou denzitometrie. Vysoký stupeň rozlíšenia sa dosahuje vďaka elektroforetickej separácii proteínov a špecifickosti monoklonálnych protilátok. Za optimálnych podmienok môže Western blotting detegovať antigén v množstvách menších ako 1 ng. Metóda sa používa na overenie pozitívnych výsledkov imunohistochemických štúdií. Pri Duchennovej svalovej dystrofii je množstvo dystrofínu 0 – 5 % normy.
Genetický krvný test . Ide v súčasnosti o najpresnejšiu metódu diagnostiky Duchennovej myodystrofie (a iných dystrofinopatií), ktorá je pre vysoký informačný obsah často jedinou doplnkovou metódou diagnostiky tohto ochorenia v zahraničí. Je to spôsobené tým, že dystrofinopatie sa vyvíjajú výlučne s mutáciami v jednom géne - géne DYS. Na štúdiu sú potrebné 2 ml venóznej krvi. Štúdia prebieha metódami hľadania delécií/duplikácií a analýzy (skenovania) mutácií, čo je pomerne časovo a finančne nákladné.
Prenatálna diagnostika Duchennovej myodystrofie je možná metódami molekulárno-genetického výskumu (analýza DNA fetálnych buniek na charakteristické mutácie), biopsiou svalov plodu a preimplantačnou génovou diagnostikou.
Liečba Duchennovej svalovej dystrofie doteraz nebola vyvinutá. Existujú len spôsoby, ako trochu spomaliť progresiu ochorenia a zlepšiť kvalitu života pacientov.
Medikamentózna terapia, ktorá ovplyvňuje priebeh ochorenia, zahŕňa:
Užívanie glukokortikosteroidov. Je dokázané, že ak sa liečba perorálnymi glukokortikosteroidmi začne v čase zastavenia telesného vývoja dieťaťa (zvyčajne vo veku 4-6 rokov), dochádza k citeľnému oneskoreniu progresie strát. svalová hmota, zvýšenie svalovej sily a zlepšenie funkčného stavu pacienta. Ak sa glukokortikosteroidy predpisujú, keď už dieťa stratilo schopnosť pohybu, ich účinnosť sa blíži k nule. Zároveň je nemožné vysvetliť účinok tejto skupiny liekov iba imunosupresiou, pretože vymenovanie imunosupresíva azatioprínu u pacientov s Duchennovou svalovou dystrofiou, ako ukázali štúdie, nie je sprevádzané žiadnym terapeutickým účinkom. Optimálny terapeutický režim prednizolón denný príjem per os v dávke 0,75 mg/kg/deň (ale nie viac ako 40 mg/deň) sa považuje za významný vedľajšie účinky, po ktorej sa dávka postupne znižuje na 0,5 mg / kg / deň av prípade pretrvávajúcich závažných vedľajších účinkov až na 0,3 mg / kg / deň. Alternatívnym režimom je užívanie rovnakých dávok každý druhý deň alebo "prerušovaný" príjem (10 dní na, 10-20 dní prestávka). Osoby, ktoré na pozadí denného príjmu prednizolón vzniká obezita a problémy so správaním, možno odporučiť prechod na 5 mg/kg dvakrát týždenne (napr. v piatok a sobotu). Pozitívny účinok (zvýšenie svalovej sily) sa pozoruje už 10. deň od začiatku liečby. V európskych a niektorých ďalších krajinách sa používa syntetický derivát prednizolónu - deflazacort(vyrobené vo Veľkej Británii, Španielsku, Indii, Brazílii, Paname a Hondurase). Ukázalo sa, že spôsobuje menej vedľajších účinkov, najmä čo sa týka obezity, avšak pri jeho užívaní dochádza častejšie k rozvoju asymptomatického šedého zákalu. Dávkovanie deflazacorta- 0,9 mg / kg / deň (ale nie viac ako 39 mg / deň). Mnohí odborníci odporúčajú ponechať pacientovi udržiavaciu dávku glukokortikosteroidov aj po jeho presune na invalidný vozík: umožňuje vám to dlhšie udržať silu v rukách, spomaliť progresiu kardiorespiračných porúch a rozvoj skoliotickej deformity chrbtice. Medzi hlavné vedľajšie účinky dlhodobej liečby glukokortikosteroidmi patria: poruchy správania, retardácia rastu, obezita, osteoporóza, porucha glukózovej tolerancie, imunosupresia, adrenálna insuficiencia, dyspepsia, peptické vredy, šedý zákal, kožné prejavy. Väčšina z nich sa dá zvládnuť bez zníženia dávky lieku.
Príjem agonistov β 2 -adrenergných receptorov. Niekoľko randomizovaných štúdií preukázalo pozitívny vplyv β 2 -agonistov na svalovú silu, neovplyvňujú však priebeh ochorenia. Tieto látky sa používajú na liečbu bronchiálnej astmy a zahŕňajú lieky ako salbutamol, formoterol atď.
Užívanie iných liekov. Možno nejaký pozitívny vplyv na priebeh ochorenia majú aminokyseliny, karnitín, koenzým-Q10, rybí tuk, extrakt zelený čaj a vitamín E.
Užívanie kardiotropných liekov. Asi 2/3 pacientov s Duchennovou myodystrofiou majú určité srdcové problémy a možno najvýznamnejším z nich je rozvoj dilatačnej kardiomyopatie. Keď má pacient svoje echokardiografické (alebo klinické) príznaky, predpisujú sa ACE inhibítory; ak po 3 mesiacoch liečby nenastane zlepšenie, pridajú sa β-blokátory (karvedilol alebo metoprolol). V prípade progresívneho priebehu sa pridávajú diuretiká, digitalisové prípravky.
nedrogové metódy. Podporujte fyzickú aktivitu: sedavý obrazživot urýchľuje progresiu svalovej dysfunkcie. Ukázané sú aj fyzioterapeutické postupy a sedenia s logopédom. Používa sa aj umelá ventilácia pľúc, rôzne ortopedické pomôcky, motorové invalidné vozíky.
Napriek všetkým týmto opatreniam je prognóza ochorenia celkovo nepriaznivá: väčšina pacientov zomiera vo veku okolo 25 rokov, najčastejšie na poruchy dýchania.
V súčasnosti vo svete prebieha výskum zameraný na vývoj nových perspektívnych metód liečby Duchennovej myodystrofie.
Génová terapia. Napriek zjavnému pokroku v štúdiách štruktúry génu dystrofínu, jeho produktov a pri objasňovaní biomechanizmov ochorenia, skutočný úspech v génovej terapii DMD ešte nebol dosiahnutý. Dôvodom je zrejme nielen obrovská veľkosť génu a jeho messenger RNA (mRNA), ale hlavne nedostatok účinnými prostriedkami dodanie génu do svalov, ako aj vývoj imunitnej odpovede na zavedenie génu. Predpokladá sa, že dosiahnutie terapeutického účinku je možné pri úspešnej transfekcii najmenej 20% a podľa najnovších údajov dokonca 40% všetkých svalových vlákien, a to nielen kostrových svalov, ale aj svalov srdca a bránice. Hlavnými kritériami účinnosti transfekcie sú zároveň: výskyt svalových vlákien pozitívnych na dystrofín, normalizácia hladiny biochemických markerov DMD, zmeny fyziologických parametrov (svalová sila atď.).
V súčasnosti aktívne vyvíjaných prístupoch génovej terapie existuje niekoľko smerov: 1) korekcia defektov zavedením normálnych kópií komplementárnej DNA (cDNA) génu dystrofínu ako súčasti rekombinantných vírusových častíc alebo nevírusovými metódami dodávania; 2) korekcia mutácií na úrovni genómovej kópie génu alebo na jeho primárnom RNA transkripte; 3) aktivácia vo svalových vláknach a bunkách autozomálneho homológu génu pre dystrofín, génu pre utrofín, potlačeného počas ontogenézy.
Transfekcia svalových vlákien pomocou vírusových vektorov . Experimenty sa uskutočňovali ako s retrovírusovými vektormi nesúcimi skrátenú cDNA "mini" génu DYS, tak s adenovírusovými vektormi schopnými niesť cDNA tohto génu plnej dĺžky. V pokusoch na myšiach bolo možné preukázať pomerne účinnú a dlhodobú transfekciu kostrového a srdcového svalstva po intravenóznom podaní rekombinantného adenovírusu s cDNA génu dystrofínu. Bola tiež preukázaná zásadná možnosť transfekcie a syntézy dystrofínu v bránicových svaloch mdx myší (laboratórna populácia myší s defektmi v géne DYS) pomocou plnej dĺžky cDNA ľudského génu pre dystrofín. Okrem normalizácie syntézy dystrofínu bolo možné preukázať, že nadmerná expresia tohto génu (hladina proteínu 50-krát vyššia ako normálne) nemá škodlivé vedľajšie účinky. Súčasne použitie vírusových nosičov, najmä v experimentoch in vivo, naráža na značné metodologické ťažkosti. Medzi ne patrí - nedostatočná kapacita balenia v retrovírusoch, potreba prítomnosti pomocných buniek. Najväčšou vážnou prekážkou použitia vírusových vektorov na dodávanie genetických konštruktov je výrazná imunitná odpoveď na vírusové antigény. Napriek obrovskému množstvu práce na modifikácii genómu nosného vírusu, zmenšeniu veľkosti vírusového genómu na najmenšiu možnú veľkosť, imunitná odpoveď napriek tomu pretrváva a opakované zavádzanie génových konštruktov je zbytočné. Napriek tomu práca na zlepšovaní metód dodávania vírusov nekončí. Najsľubnejšie je zavedenie génu dystrofínu do novonarodených myší. Ukázalo sa, že v dôsledku transfekcie myší adenovírusovým vektorom a zhutnenia DNA polylyzínom pK8 sa expresia dystrofínu zaznamenávala takmer 1 rok.
Nevírusové spôsoby dodania cDNA génu dystrofínu . Metódy nevírusového podávania zahŕňajú balistickú transfekciu, metódy elektroporácie (elektrošok), zavedenie genetických konštruktov do zloženia lipozómov alebo zbalených s oligopeptidmi, molekulárnymi konjugátmi, polymérnymi nosičmi. Tieto nosiče do značnej miery nemajú nevýhody vlastné vírusovým vektorom, avšak schopnosť transformácie vo väčšine z nich je nižšia ako schopnosť vírusových vektorov. Prvé experimenty s dodaním „nahej“ plazmidovej DNA s cDNA ľudského génu pre dystrofín ukázali možnosť transfekcie a objavenie sa svalových vlákien pozitívnych na dystrofín u mdx myší.
Doposiaľ najpokročilejšie sú štúdie o prenose dystrofínového génu elektroporáciou alebo s nosičom na báze polymérnej formy dextránu. V druhom prípade sa na dodanie génu dystrofínu použil dextrán, čím sa vytvoril samoskladajúci sa komplex DNA polyméru. Absencia toxicity a imunitnej odpovede, diseminácia do rôznych svalových skupín a dostatočne dlhodobá (viac ako dva mesiace) expresia ukázali prísľub tohto aplikačného systému pre klinické skúšky.
Ešte povzbudzujúcejšie výsledky sa získali v experimentoch na myšiach, potkanoch, králikoch a opiciach pri dodávaní genetických konštruktov do svalov pomocou elektroporácie. Osemnásobný elektrický impulz (200 V/cm2, 20 ms, 17 Hz), 30 sekúnd po zavedení plazmidov s génom LacZ β-galaktozidázy, viedol k syntéze β-galaktozidázy v 76 % svalových vlákien, a s použitím elektrického šoku len v 8 %.
Génová terapia na úrovni primárneho transkriptu génu pre dystrofín . Z týchto metód priťahuje zvláštnu pozornosť technika cielenej straty exónu nesúceho mutantný stop kodón, vyvinutá v laboratóriu Georgea Dixona vo Veľkej Británii. Práca bola vykonaná in vitro na myoblastoch mdx myší s nezmyselnou mutáciou v exóne 23 génu dystrofínu. V podmienkach in vitro sa ukázalo, že už 6 hodín po transfekcii špecifickými oligonukleotidmi (antisense oligonukleotidy) došlo k odstráneniu mutantného exónu 23 u 50 % mRNA a po 24 hodinách u 100 % mRNA. Prísľub tohto prístupu spočíva v tom, že myoblasty začnú syntetizovať dystrofínový proteín plnej dĺžky, hoci je defektný v jednom funkčne nevýznamnom exóne. Po transplantácii pacientovi budú modifikované myoblasty schopné obnoviť funkciu a zabrániť smrti postihnutých svalových vlákien.
Vzhľadom na originalitu metódy a jej veľkú perspektívu, zastavme sa pri krátkom vysvetlení jej podstaty. Pripomeňme si, že dystrofínový proteín vykonáva funkciu „kotvy“, ktorá spája proteíny „kostra“ svalového vlákna so štruktúrami spojivového tkaniva obklopujúcimi svalové vlákno. Slúži ako "tlmič otrasov", ktorý chráni svalové vlákna pred poškodením pri svalovej kontrakcii a relaxácii. Schématicky môže byť dystrofín znázornený ako lano spájajúce kotvu a loď. Kotva môže plniť svoju funkciu len vtedy, ak je spojená s lanom člna.

Schematické znázornenie "kotvovej" funkcie dystrofínu (
Siedmy september je Medzinárodným dňom povedomia o Duchennovom syndróme. Ide o jednu z najčastejších dedičných genetických patológií – diagnostikuje sa u jedného z 3 000 novorodencov. Doteraz neexistuje spôsob, ako chorobu úplne vyliečiť, no nový vývoj ponúka šancu situáciu zmeniť. Aká by mohla byť terapia budúcnosti, prezradí MedAboutMe.
Čo je Duchennova svalová dystrofia?
Duchennova svalová dystrofia je dedičná genetická porucha, jedna z tých, ktoré postihujú iba chlapcov. Choroba sa prenáša od matiek, no samotné ženy ňou netrpia, ale sú zdravými prenášačkami postihnutého génu. Gén sa môže prenášať po ženskej línii po mnoho generácií a neprejavuje sa, takže narodenie dieťaťa s DMD je pre rodinu často prekvapením.
Patológia spočíva v géne kódujúcom proteín dystrofín - v prípade choroby sa produkuje v nedostatočnom množstve alebo úplne chýba. A keďže dystrofín je základom svalových vlákien, u detí s DMD svaly postupne ochabujú, degenerujú a sú nahradené tukovým alebo spojivovým tkanivom. To vedie k invalidite - choroba neustále postupuje, postupne zachytáva viac a viac svalov.
Aj pri súčasnej liečbe sa chlapci s Duchennovou svalovou dystrofiou dožívajú v priemere 20-25 rokov. V niektorých prípadoch sa pacienti dožívajú 40 rokov, no zatiaľ je to stále skôr výnimka ako pravidlo.
Prvé príznaky ochorenia sa vyskytujú pred dosiahnutím veku 3 rokov, v tomto veku sa u dieťaťa prejavuje:
rýchla únavnosť; oneskorenie vo vývoji; ťažkosti pri zvládnutí zručnosti chôdze, deti často chodia po prstoch; Gowersov príznak - keď sa dieťa snaží vstať, aktívne si pomáha rukami, pretože svaly na nohách už nezvládajú.
Choroba postupuje a už u pacientov vo veku 15-18 rokov sa pozorujú tieto príznaky:
deformácia kostry; neschopnosť pohybovať sa bez invalidného vozíka, postaviť sa, samostatne sedieť, niekedy dokonca hýbať rukami; endokrinné poruchy sa pozorujú u 30-50% pacientov; poškodenie srdcového svalu, kardiomyopatia; poškodenie dýchacích svalov; niekedy dochádza k narušeniu duševného vývoja.
V štádiu, keď choroba zachváti srdce a pľúca, je už takmer nemožné pacientovi pomôcť.
Štandardné liečby
Chorobu opísal ešte v 19. storočí neurológ Guillaume Duchenne, po ktorom je pomenovaná. A od tej doby lekári hľadajú efektívnymi spôsobmi terapiu. Prvou metódou, ktorá sa používa dodnes, bolo použitie ortopedických pomôcok.
Svalový tréning pomáha mierne spomaliť proces ich ničenia. Pre pacientov sa využíva aktívny (kým sú schopní cvičiť) aj pasívny tréning. Lekári poznamenávajú, že pravidelné cvičenie môže predĺžiť obdobie, keď sa pacient pohybuje bez invalidného vozíka.
Podľa štatistík takmer polovica pacientov s myodystrofiou podstúpi operáciu chrbtice. Pri slabom svalstve je kostra ohnutá, čo výrazne zhoršuje priebeh ochorenia. Preto takýmto pacientom prichádzajú na pomoc rôzne ortopedické pomôcky – fixácia kostí a kĺbov im umožňuje spomaliť ich deformáciu.
Moderný vývoj v tejto oblasti terapie už dávno presahuje rámec jednoduchých podporných prostriedkov. Napríklad robotické rameno A-Gear vyvinuté holandskou univerzitou v Twente je schopné nahradiť atrofovanú končatinu u ľudí. umelé rameno zachytáva elektrické signály zo svalov alebo minimálne svalové napätie. Ľudia s Duchennovou svalovou dystrofiou dokážu ovládať mechanickú končatinu, pričom pohyby sú veľmi presné a prirodzené. "Do štúdie sme zaradili veľa účastníkov, ktorí už stratili schopnosť pohybovať sa rukami na 3-5 rokov," hovorí výskumník Joan Lobo-Prat. "A-Gear im pomohol obnoviť niektoré funkcie končatín."
Lieky na Duchennovú svalovú dystrofiu
Medikamentózna liečba je dôležitou súčasťou všeobecnej terapie. Najobľúbenejšou skupinou liekov u pacientov s Duchennovou svalovou dystrofiou sú steroidy: prednizolón, deflazacort a iné. Začínajú ich užívať vo veku 4-6 rokov, kedy sa prejavia príznaky ochorenia, no nedochádza k výraznému zhoršeniu stavu. Steroidy pomáhajú oddialiť rozvoj závažných symptómov, ale z dlhodobého hľadiska nie sú účinné. Len čo atrofia u pacienta začne postupovať, tieto lieky už nedokážu zlepšiť stav svalov. Napriek tomu steroidy zostávajú dôležitou súčasťou liečby DMD.
Existuje aj ďalší vývoj zameraný na zlepšenie výkonu svalov. Takže v roku 2016 FDA (US Food and Drug Administration) schválil liek Exondys51 (etiplirsen), ktorý sa môže stať konkurentom steroidov. Podľa štúdií je látka schopná posilniť svalové tkanivo, pretože zvyšuje hladinu dystrofínu v kostrových svaloch. Nie všetci lekári však zdieľajú názor FDA, pretože schválenie lieku prebehlo v zrýchlenom konaní. V skutočnosti v súčasnosti neexistuje žiadny jednoznačný dôkaz, že môže oddialiť rozvoj paralýzy alebo zmierniť príznaky pokročilého ochorenia. To znamená, že Exondys51, podobne ako steroidy, je účinný len v krátkom období choroby. Napriek tomu, že liek už bol schválený, FDA pokračuje vo výskume. Ak sa účinnosť lieku nepotvrdí, jeho výroba sa zastaví.
Európsky výbor pre lieky na humánne použitie (CHMP) zaregistroval v roku 2014 ďalší liek – Translarna (ataluren). Na rozdiel od iných liekov, ktoré iba posilňujú svaly, ataluren je schopný obnoviť narušenú syntézu bielkovín. Takže teoreticky eliminuje samotnú príčinu vývoja choroby. Pretože to nový vývoj, je stále ťažké pochopiť, ako efektívne je to z dlhodobého hľadiska. Ale priebežné výsledky sú celkom povzbudivé.
Úprava genómu a technológia CRISPR/Cas Duchennova svalová dystrofia je spôsobená abnormalitou v géne, takže akákoľvek medikamentózna alebo fyzioterapeutická liečba je len symptomatická. Človeka možno vyliečiť iba pomocou génovej terapie – úpravou kódu DNA a zmenou „chorého“ génu. Napriek tomu, že doteraz sa takáto liečba neuplatňovala, nové technológie ponúkajú šancu na vyriešenie problému v budúcnosti.
Technológia CRISPR/Cas9 si v poslednej dobe získala mimoriadnu popularitu. Je založený na pôsobení špeciálneho enzýmu Cas9. Preniká do bunky a tam je schopný konkrétny úsek reťazca DNA nielen rozpoznať, ale aj odstrániť. Keďže lekári vedia, ktorý gén je zodpovedný za vznik ochorenia a kde presne sa nachádza, pomocou techniky môžu jednoducho vyrezať kúsok DNA s patológiou. A to prinesie úplné zotavenie.
Nedávne štúdie však odhalili množstvo ťažkostí s používaním tohto systému. Po pokuse na embryách sa ukázalo, že metóda naozaj umožnila vyliečiť až 30 % buniek. Ale zároveň takýto úvod vyvolal veľký počet ďalších mutácií.
Možnosti CRISPR/Cas9 aktívne skúmajú mnohé laboratóriá po celom svete. Pozitívne výsledky zaznamenali najmä vedci z Southwestern Medical Center Texaskej univerzity. Tu sa CRISPR/Cas9 úspešne používa na liečbu myší s Duchennovou svalovou dystrofiou. Bola vyvinutá aj presnejšia technológia vhodná práve pre toto ochorenie – v nej bol proteín Cas9 nahradený proteínom Cpf1. "Odobrali sme bunky od pacientov s DMD a dokázali sme ich opraviť v laboratóriu," hovorí Dr. Eric Olson. "Po aplikácii CRISPR/Cpf1 sa v bunkách obnovila produkcia dystrofínu."
Iný výskum: Umelý chromozóm, vírusy a mutácie
Skupina japonských a britských vedcov navrhla ďalší alternatívny prístup k liečbe DMD - zavedenie umelých zdravých chromozómov (gén s patológiou sa nachádza na X chromozóme 23. páru). Takýto chromozóm sa umiestni do kmeňovej bunky a neskôr sa zavedie do tela a pomôže obnoviť svaly. Pozitívne výsledky sa zatiaľ dosiahli pri pokusoch na myšiach. Podobné štúdie sa uskutočnili v Rusku, v r.
Ďalší možný smer v liečbe DMD sa objavil vďaka psovi. Pred viac ako desiatimi rokmi vedci z University of São Paulo v Brazílii chovali šteniatka s svalová dystrofia Duchenne. Napriek diagnóze potvrdenej testami sa však u jedného z nich, psa Ringa, choroba neprejavila, navyše zviera žilo 11 rokov bez akýchkoľvek príznakov. Vedci to pripisujú prítomnosti inej patológie u psa – mutácie v géne Jagged1. "Nevieme presne, aká koncentrácia proteínu Jagged1 by mala byť vo svaloch, aby účinne chránila pred rozvojom ochorenia," hovorí genetik Louis Kunkel. "Ale Ringov príbeh naznačuje, že mutácia Jagged1 kompenzuje atrofiu spôsobenú nedostatkom dystrofínu."
Jednou z najsľubnejších vyhliadok je rozsiahla štúdia zahŕňajúca skutočných pacientov. Podstatou experimentu je pokúsiť sa zaviesť do bunky opravenú kópiu génu pre dystrofín. A plánuje sa to urobiť pomocou vírusov - stanú sa nosičmi požadovaného fragmentu DNA. Testy sa uskutočnia v Spojených štátoch už koncom roka 2017 alebo začiatkom roka 2018. Na výskume sa bude podieľať najmä farmaceutická spoločnosť Pfizer. Doposiaľ bolo v Ohio's Nationwide Children's Hospital vybratých 12 detí rôznych vekových skupín, ktorým boli vpichnuté vírusové častice.
Vrodená slabosť svalov, ktorá postupuje s vývojom tela, sa v medicíne nazýva „Duchennova myodystrofia“. Toto ochorenie sa vyskytuje výlučne u mužov. Pochopenie podstaty patológie umožňuje zmierniť jej priebeh a pomôcť deťom vyrovnať sa s počiatočnými prejavmi.
Charakteristika ochorenia
Duchennova svalová dystrofia je genetická patológia spôsobená porušením štruktúry svalových vlákien. Postupne sa rozpadajú a človek stráca schopnosť pohybu. Ochorenie sa prejavuje už v dojčenskom veku. Najprv dochádza k poruchám svalov, potom sa pripájajú deformity kostry. Klinický obraz dopĺňajú endokrinné a duševné poruchy.
Prvýkrát bola myodystrofia opísaná v roku 1861 francúzskym neurológom, ktorého meno bolo neskôr pomenované. Je diagnostikovaná pomerne často: 1 prípad na 3500 novorodencov. Neexistuje žiadny radikálny liek. Terapia, ktorú ponúkajú lekári, je výlučne symptomatická. Pacienti s touto diagnózou len zriedka prežijú míľnik 30 rokov.
Hlavné dôvody
Svalová dystrofia je výsledkom odchýlky v genetický kód DNA. Mutácia sa vyskytuje v géne umiestnenom na X chromozóme. Jedna z jeho sekcií je zodpovedná za produkciu špeciálneho proteínu – dystrofínu. Táto látka na mikroskopickej úrovni tvorí základ svalových vlákien a plní niekoľko funkcií:
- udržiavanie bunkovej kostry;
- zabezpečenie schopnosti svalových vlákien sťahovať sa a relaxovať.
Pri tejto chorobe dystrofín chýba alebo je slabo syntetizovaný. Hladina "normálnych" bielkovín nepresahuje 3%. Táto mutácia vedie k deštrukcii vlákien vo svaloch. Postupne sa znovuzrodia, nahradí ich tukové a spojivové tkanivo. V dôsledku toho človek stráca schopnosť pohybu.
Aký typ dedičnosti má Duchennova svalová dystrofia? Ochorenie sa prenáša recesívnym spôsobom. V ľudskom tele sú všetky gény spárované. Aby sa pri dedičnom ochorení objavili patologické poruchy, musí sa genetický defekt vyskytnúť v jednom chromozóme alebo v podobných častiach oboch. V druhom prípade hovoríme o recesívnom type dedenia.
Ak je genetická chyba diagnostikovaná iba v jednom chromozóme, ale choroba postupuje, hovoria o dominantnom znaku prenosu. Recesívny typ je možný so súčasnou léziou rovnakých štruktúr DNA. Keď je druhý chromozóm absolútne "zdravý", patológia sa nevyvíja. Preto je dystrofia diagnostikovaná iba u mužov. Vo svojej genetickej sade majú jeden chromozóm X a druhý (Y) je pár.
Čo hovorí veda o nežnom pohlaví? Duchennova svalová dystrofia je u dievčat zriedkavá. Aby to bolo možné, musia sa dva patologické chromozómy X v genotype zhodovať, čo je nepravdepodobné. Dievčatá môžu pôsobiť len ako prenášačky choroby a prenášať ju na svojich synov.

Všeobecný klinický obraz
Ochorenie zanecháva stopy na nervovosvalovom systéme. Jeho prejavy možno pozorovať u detí vo veku 2-3 rokov. Rodičia si začínajú všímať, že dieťatko zaostáva fyzický vývoj od rovesníkov. Patologický proces rýchlo postupuje a šíri sa do dolných končatín. Potom sa presunie do iných častí svalov.
Poškodenie svalového korzetu a nadmerné zaťaženie vedú k zakriveniu končatín. U chorých detí dochádza aj k zmenám vo fungovaní srdca, mentálnej retardácii.
Všetky patologické prejavy ochorenia možno rozdeliť do niekoľkých skupín:
- poškodenie kostrového svalstva;
- poruchy v práci srdca;
- deformácia kostry;
- mentálne poruchy;
- endokrinné poruchy.
Zvážte klinické prejavy každej zo skupín podrobnejšie.
Poranenie kostrového svalstva
Deti sa rodia bez vážnejších zdravotných problémov. Po niekoľkých mesiacoch však ich motorický vývoj začne zaostávať. Tieto deti sú menej aktívne. Lekári a rodičia si stále nevšimnú zjavné odchýlky, pripisujú všetko zvláštnostiam temperamentu.
Počiatočné príznaky ochorenia sa vyskytujú po prvých krokoch. Deti s DMD neustále padajú a chodia po špičkách. Ak je väčšina rovesníkov už sebavedomá na nohách, tvrdohlavo naďalej pociťujú ťažkosti s pohybom.
Ďalšou fázou prejavu choroby je obdobie, keď deti nadobúdajú schopnosť rozprávať. Začnú sa sťažovať rodičom na slabosť a únavu. Skákanie na ihrisku, beh, lezenie na hrazdách – všetky tieto aktivity im neprinášajú potešenie.
Aké sú ďalšie príznaky Duchennovej svalovej dystrofie? Gowerova porucha sa považuje za zvláštny prejav choroby. Pri každom pokuse vstať z podlahy si dieťa rukami takto pomáha slabým svalom nôh. Za týmto účelom si o seba opiera končatiny a triedi ich po celom tele.
Postupná progresia ochorenia vedie k tomu, že choré deti do 10-12 rokov strácajú schopnosť samostatného pohybu. Väčšina z nich potrebuje invalidný vozík. Schopnosť udržať telo vo vzpriamenej polohe trvá len do 16 rokov.

Deformácie kostry
Táto skupina zahŕňa symptómy spojené so svalovými poruchami. Duchennova svalová dystrofia sa prejavuje zväčšením bedrového zakrivenia, ktoré je doplnené zakrivením hrudnej chrbtice a zhrbením. Mnoho detí mení tvar chodidla. Postupom času sa rozvinie osteoporóza. Tieto príznaky sa ďalej zhoršujú klinický obraz prispievajú k zhoršovaniu pohybových porúch.
Poruchy v práci srdca
Progresívna svalová dystrofia je nevyhnutne sprevádzaná poškodením srdcového svalu. Spravidla sa vyvíja kardiomyopatia. Klinicky sa prejavuje poklesom tlaku, porušením srdcového rytmu. Hranice hlavného svalu tela sa zvyšujú, ale zároveň sa výrazne znižuje jeho funkčnosť. Výsledkom je zlyhanie srdca.
Kombinácia týchto defektov s respiračnou dysfunkciou často spôsobuje smrť.
mentálne poruchy
Tento príznak sa považuje za voliteľný, ale možný. Jeho vzhľad môže byť spôsobený nedostatkom jednej z odrôd dystrofínu - apodystrofínu, ktorý sa nachádza v mozgu. Zároveň slabé svaly a závažnosť duševných porúch nie sú navzájom prepojené. Neschopnosť dieťaťa navštíviť MATERSKÁ ŠKOLA a škola len posilňuje kognitívne poruchy.

Endokrinné poruchy
U 30 – 50 % pacientov sú diagnostikované rôzne druhy endokrinných porúch. Môžu byť vyjadrené vo forme obezity alebo nedostatočného rozvoja pohlavných orgánov. Nadmerné usadeniny sa najčastejšie pozorujú v oblasti mliečnych žliaz, ramenného pletenca a zadku. Pacienti s Duchennovou dystrofiou bývajú nízkeho vzrastu.
lekárska prehliadka
Diagnóza Duchennovej myodystrofie je založená na niekoľkých typoch štúdií, z ktorých hlavnou je test DNA. Detekcia defektu na X chromozóme v oblasti zodpovednej za syntézu dystrofínu sa považuje za konečné potvrdenie diagnózy.
Medzi ďalšie diagnostické metódy, ktoré sa dnes používajú:
- CPK (stanovenie aktivity kreatínfosfokinázy). Tento enzým je priamym odrazom odumierania svalových vlákien. U detí s Duchennovou dystrofiou jej miera stonásobne prekračuje normu.
- Elektromyografia.
- Dychové testy, EKG, ultrazvuk srdca. Umožňuje identifikovať odchýlky v práci iných orgánových systémov.
- Svalová biopsia. Pomocou tejto metódy sa stanoví obsah dystrofínu v tele.
Progresívna svalová dystrofia u dieťaťa znamená, že v genotype matky je prítomný patologický chromozóm X. Len v ojedinelých prípadoch je žena úplne zdravá. Prítomnosť defektného génu predstavuje hrozbu pre ďalšie tehotenstvá. Preto sa takýmto rodinám odporúča navštíviť genetiku.
Na začiatku opakované tehotenstvo párom sa ponúka takzvaná prenatálna diagnostika. Znamená to štúdium genotypu dieťaťa v maternici. Takáto štúdia eliminuje riziko dedičných ochorení, medzi ktoré patrí Duchennova svalová dystrofia.
Prenatálna diagnostika je založená na použití bunkového materiálu. Získava sa rôznymi postupmi: biopsia chorionu, amniocentéza atď. Uvedené manipulácie prinášajú určité riziko pre plod, ale dajú sa použiť na diagnostiku genetického ochorenia so 100% zárukou.

Lekárske ošetrenie
Duchennova svalová dystrofia je nevyliečiteľné ochorenie. Ale pacienti s takouto diagnózou by nemali zostať pripútaní na lôžko. Ako pomôcť dieťaťu predĺžiť obdobie fyzickej aktivity ponúka moderná medicína niekoľko spôsobov.
Z liekov na tento účel sú pacientom predpísané steroidy a beta-agonisty. Použitie druhého ("Albuterol", "Formoterol") nemá spoľahlivé rozpoznanie. Preto dnes nie je potrebné hovoriť o ich účinnosti. Takéto lieky sa používajú výlučne ako experimentálna liečba.
Základom terapie sú steroidy. Ich pravidelné používanie umožňuje na chvíľu doplniť svalovú silu. Lekári naznačujú, že takéto lieky môžu spomaliť progresiu ochorenia, ako aj zabrániť výskytu skoliózy. Možnosti steroidov sú však obmedzené. Duchennova svalová dystrofia sa bude aj tak ďalej rozvíjať.
Okrem toho sú pacientom predpísané lieky na srdce. Ide predovšetkým o ATP inhibítory, antiarytmiká a metabolické lieky. Umožňujú vám odolávať kardiologickým aspektom ochorenia.

Fyzioterapia a ortopedická starostlivosť
Neliečivá terapia zahŕňa vymenovanie fyzioterapie a ortopedickej starostlivosti. V prvom prípade hovoríme o rôznych masážnych technikách a plávaní. Fyzioterapeutický účinok pomáha udržať pohyblivosť a pružnosť kĺbov na dlhší čas. Mierna aktivita priaznivo ovplyvňuje priebeh ochorenia. Na druhej strane nečinnosť a pokoj na lôžku môžu klinický obraz len zhoršiť. Preto lekári odporúčajú čo najdlhšie udržiavať fyzickú aktivitu.
Ortopedická starostlivosť je dôležitou súčasťou terapie u pacientov s diagnózou Duchennovej svalovej dystrofie. Liečba liekmi spojená so špeciálnymi prístrojmi im môže výrazne uľahčiť život. Ich zoznam je veľmi rôznorodý: rôzne vertikalizátory, zariadenia na nezávislé prijatie pohodlnej polohy, elektrické invalidné vozíky, korzety na chrbticu, dlahy na nohy a mnoho ďalšieho.

Prognóza pre pacientov
Duchennova myodystrofia je závažné genetické ochorenie, ktorého prvé príznaky sa vyskytujú u detí v prvých mesiacoch života. Chlapci majú spočiatku ťažkosti s chôdzou, potom sa nevedia jednoducho postaviť z podlahy. Medikamentózna liečba steroidmi výrazne mení priebeh patologického procesu. Lieky pomáhajú na chvíľu obnoviť svalovú silu.
Je ťažké presne predpovedať, kedy pacient začne používať invalidný vozík. Zvyčajne sa potreba tohto zariadenia objavuje vo veku 8-11 rokov. S ďalším rozvojom svalovej slabosti je pre pacienta ťažké udržať polohu tela, môžu vzniknúť komplikácie.
Aká je prognóza lekárov pri diagnóze Duchennovej svalovej dystrofie? Ochorenie môže výrazne skrátiť dĺžku života. V súčasnosti sa však väčšina mladých mužov dožíva dospelosti, avšak za predpokladu kvalitnej lekárskej a fyzioterapeutickej starostlivosti.
Duchennova svalová dystrofia je zriedkavý stav. Jeho ďalší názov je Duchennova svalová dystrofia alebo progresívna Duchennova svalová dystrofia. Názov je spôsobený tým, že choroba postupuje rýchlo. Ochorenie sa vyskytuje asi u 3 ľudí zo 100 000. Patológia je spôsobená vrodenou genetickou anomáliou, je závažná a postihuje veľkú skupinu svalov. V priebehu času vedie dystrofia svalového systému k úplnej neschopnosti samostatného pohybu.
Duchennova myodystrofia vedie k patológii iných orgánov, čo výrazne znižuje dĺžku života človeka.
Prevažnú väčšinu pacientov s Duchennovou svalovou dystrofiou tvoria chlapci. Dievčatá trpia touto chorobou veľmi zriedkavo. Ide o vrodené ochorenie, ktoré je spôsobené zmeny na X chromozóme. Na chromozóme X sa nachádza gén, ktorý riadi produkciu proteínu dystrofínu. Tento proteín ovplyvňuje celistvosť membrán svalových vlákien (sarkolemy) a odolnosť svalov voči naťahovaniu. Tiež kontroluje hladiny vápnika vo svalovom tkanive a rast svalov. Ak sa v ľudskom tele vytvorí nedostatok proteínu dystrofínu, vedie to k postupnej deštrukcii svalových buniek (myocytov). Vo svaloch vznikajú degeneratívne zmeny, svalové vlákna atrofujú, rozpadávajú sa a sú nahradené tukovým a spojivovým tkanivom.
Pri progresívnej Duchennovej svalovej dystrofii obsah normálneho dystrofínu prudko klesá v dôsledku génovej mutácie. Tento proteín buď úplne chýba, alebo telo obsahuje defektný dystrofín. U chorých ľudí nie je hladina normálneho dystrofínu v tele vyššia ako 3%.
 Dievčatá a ženy veľmi zriedka trpia týmto typom svalovej dystrofie. Ale často sú nositeľmi zmeneného génu. Je to spôsobené tým, ako sa choroba prenáša cez X chromozóm. Ako viete, chromozómová sada muža je XY a ženy - XX. Ak má matka chlapca vo svojom genetickom súbore defektný chromozóm X, potom sa chlapec môže narodiť chorý, aj keď otec chorý nie je.
Dievčatá a ženy veľmi zriedka trpia týmto typom svalovej dystrofie. Ale často sú nositeľmi zmeneného génu. Je to spôsobené tým, ako sa choroba prenáša cez X chromozóm. Ako viete, chromozómová sada muža je XY a ženy - XX. Ak má matka chlapca vo svojom genetickom súbore defektný chromozóm X, potom sa chlapec môže narodiť chorý, aj keď otec chorý nie je.
Dievčatko sa narodí s Duchennovou svalovou dystrofiou iba vtedy, ak je matka nositeľkou defektného génu a otec trpí touto chorobou. Takéto prípady sú veľmi zriedkavé. Najčastejšie sa nositeľkou choroby stáva aj dievča narodené matke, ktorá je nositeľkou defektného génu a prenáša ju na svojich synov.
Progresívna DMD sa však nemusí nevyhnutne prenášať na dieťa od rodičov. Existujú prípady, keď dôjde k genetickému zlyhaniu v dôsledku náhodnej mutácie. Stáva sa tiež, že choré dieťa sa narodí absolútne zdravým rodičom, ktorí nie sú nositeľmi defektných génov.
Príznaky Duchennovej svalovej dystrofie
Zvyčajne sa choroba prejavuje vo veku 1-5 rokov. Ovplyvňuje nielen svaly kostry, ale aj iné orgány.
- Poškodenie kostrového svalstva je skoré znamenie ochorenie, ktoré sa vyskytuje u malého dieťaťa.
- Progresívna svalová slabosť.
- V dôsledku poškodenia svalov dochádza k deformácii kostí kostry.
- Ochorenie postihuje nielen svaly kostry, ale vedie aj k zmenám na srdci.Niektoré deti s Duchennovou svalovou dystrofiou zaostávajú v duševnom vývoji.
- Choroba vedie k narušeniu činnosti endokrinných žliaz.
Poranenie kostrového svalstva
Poškodenie svalov je hlavným a skorým príznakom ochorenia. Svalové symptómy sú viditeľné vo veku 1-5 rokov.
V dojčenskom veku dieťa vyzerá zdravo. Možno si len všimnúť, že takéto deti do jedného roka sú neaktívne a neradi robia akékoľvek pohyby. Najčastejšie tomu rodičia nepripisujú žiadny význam a nízku pohybovú aktivitu dieťaťa spájajú s individuálnymi vývinovými charakteristikami.

Choroba postupuje, mnohé deti strácajú schopnosť chodiť do 12 rokov. Musia používať invalidný vozík.
V dospievaní sa do bolestivého procesu zapájajú dýchacie svaly. Pre dieťa sa stáva ťažké dýchať, je rušené astmatickými záchvatmi, najmä v noci. Z tohto dôvodu sa deti boja spať. To môže viesť k zlyhaniu dýchania.
Kostné lézie
Zmeny vo svaloch vedú k poškodeniu kostí kostry. Existujú zakrivenie chrbtice (skolióza, lordóza), zhrnutie (kyfóza). A tiež hrudník a chodidlá sú ohnuté. Kosti sa stávajú tenšie a krehkejšie (difúzna osteoporóza). Poškodenie kostí ďalej obmedzuje schopnosť pacientov pohybovať sa samostatne.
Poruchy srdca
 Duchennova svalová dystrofia spôsobuje kardiomyopatiu. Do patologického procesu sa zapája aj srdcový sval. Srdce sa zväčšuje, zatiaľ čo jeho funkcie sú narušené. Pacienti sa sťažujú na arytmiu a skoky v krvnom tlaku. V priebehu času sa môže vyvinúť srdcové zlyhanie.
Duchennova svalová dystrofia spôsobuje kardiomyopatiu. Do patologického procesu sa zapája aj srdcový sval. Srdce sa zväčšuje, zatiaľ čo jeho funkcie sú narušené. Pacienti sa sťažujú na arytmiu a skoky v krvnom tlaku. V priebehu času sa môže vyvinúť srdcové zlyhanie.
Hormonálne poruchy
Duchennova svalová dystrofia často vedie k rozvoju Cushingovho syndrómu. K obezite dochádza pri ukladaní tuku v hornej časti tela. Obezita je kombinovaná s nedostatočnou funkciou gonád. Niekedy sú pohlavné orgány nedostatočne vyvinuté. Pacienti s Duchennovou svalovou dystrofiou sú často nízkej postavy a majú nadváhu.
Intelektuálne postihnutie
Mentálna retardácia nie je pozorovaná vo všetkých prípadoch. Približne 30 % pacientov s Duchennovou myodystrofiou má mentálnu retardáciu a nízke IQ. Je to spôsobené nedostatkom apodystrofínu v mozgu. Celkový nedostatok dystrofínového proteínu v tele vedie k nedostatku jeho špeciálnej formy – apodystrofínu. Táto látka je nevyhnutná pre normálne fungovanie mozgu, jej nedostatok vedie k duševným poruchám. Stupeň mentálnej retardácie pri tomto ochorení nijako nesúvisí so závažnosťou svalových porúch. Pri silnej svalovej slabosti môže existovať normálna inteligencia.
Kvôli neschopnosti normálneho pohybu sú takéto deti často izolované od spoločnosti svojich rovesníkov, nemôžu navštevovať predškolské a školské zariadenia. To môže zhoršiť duševné poruchy.
Diagnóza Duchennovej myodystrofie
Na diagnostiku myodystrofie sa používa niekoľko typov výskumu:

Liečba Duchennovej svalovej dystrofie
K dnešnému dňu neexistuje radikálna liečba tejto choroby. Choroba sa považuje za nevyliečiteľnú a progresívnu. Nevyhnutne vedie k invalidite pacienta.
Na zmiernenie prejavov ochorenia je možná symptomatická terapia.
Lieky na liečbu Duchennovej myodystrofie

Fyzioterapia
Fyzioterapeutické metódy liečby pomáhajú na chvíľu zachovať motorickú funkciu pacienta. Pacienti sú kontraindikovaní v úplnej nehybnosti a odpočinku na lôžku, to vedie len k veľmi rýchlemu rozvoju ochorenia. Pacienti potrebujú miernu aktivitu.
- Užitočné sedenia masáže a cvičebnej terapie.
- Aby sa normalizovala funkcia dýchania, sú zobrazené dychové cvičenia.
Ortopedická starostlivosť
 Ak dôjde k strate motorických funkcií v dôsledku choroby, musia sa použiť ortopedické pomôcky. Pri svalových kontraktúrach sa používajú ortézy a špeciálne dlahy. Ak sa vytvorilo vážne zakrivenie chrbtice, potom pomáha použitie korzetov. Pri úplnej nemožnosti samostatného pohybu a státia sa používajú vertikalizátory a elektrické invalidné vozíky.
Ak dôjde k strate motorických funkcií v dôsledku choroby, musia sa použiť ortopedické pomôcky. Pri svalových kontraktúrach sa používajú ortézy a špeciálne dlahy. Ak sa vytvorilo vážne zakrivenie chrbtice, potom pomáha použitie korzetov. Pri úplnej nemožnosti samostatného pohybu a státia sa používajú vertikalizátory a elektrické invalidné vozíky.
Vývoj nových liečebných postupov
Ani pri použití všetkých moderných metód liečby nie je možné úplne poraziť Duchennovú svalovú dystrofiu. Predpokladaná dĺžka života pacientov je veľmi krátka. Preto prebieha výskum nových liečebných metód.
- Skúma sa možnosť nahradenia defektného génu zdravým génom.
- Liečba kmeňovými bunkami je predmetom skúmania.
- Prebieha výskum transplantácie buniek schopných produkovať proteín dystrofín.
- Na zvieratách sa uskutočňujú pokusy s cieľom nahradiť proteín dystrofín utrofínom.
- Skúma sa možnosť spomalenia ochorenia korekciou génu (preskočenie exónu).
Prognóza a prevencia ochorenia
K dnešnému dňu je prognóza Duchennovej svalovej dystrofie nepriaznivá. Choroba postupuje a vedie k smrti. Väčšina pacientov sa nedožije veku 20-30 rokov. Smrť nastáva v dôsledku zlyhania srdca a dýchania a súvisiacich infekcií.
Prenatálna diagnostika zohráva dôležitú úlohu v prevencii chorôb. Ak už má rodina dieťa s Duchennovou svalovou dystrofiou, potom to vo väčšine prípadov znamená, že matka je nositeľkou defektného X chromozómu. A to znamená, že v ďalších tehotenstvách existuje riziko chorého dieťaťa. Preto je potrebné poradiť sa s genetikom a vykonať prenatálne štúdie (amniocentéza, choriová biopsia). Tieto metódy dokážu presne určiť, či má plod genetickú poruchu.